Leidimo atlikti klinikinį vaistinio preparato tyrimą ir (ar) esminį klinikinio vaistinio preparato tyrimo pakeitimą išdavimas (pagal Reglamento (ES) Nr. 536/2014 reikalavimus)
Klinikinio vaistinio preparato tyrimo paraiškos vertinimo Lietuvos bioetikos komitete schema
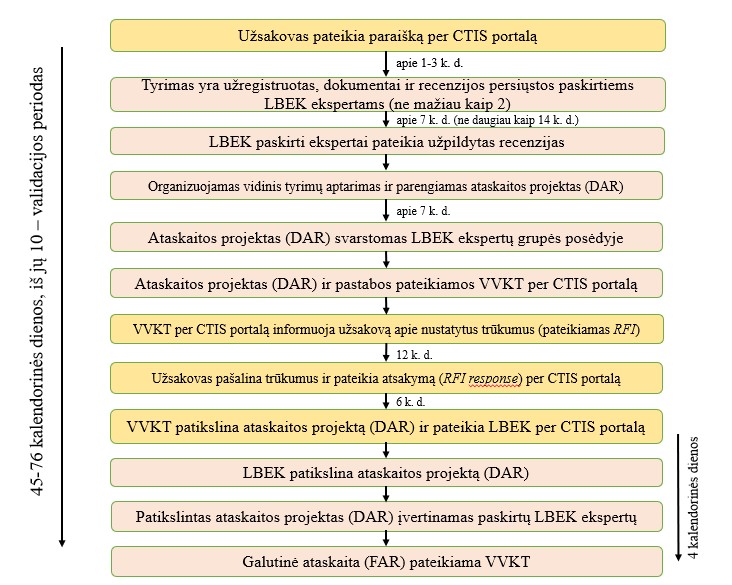{kind=link}
Klinikinio vaistinio preparato tyrimo paraiškos vertinimo CTIS portale schema
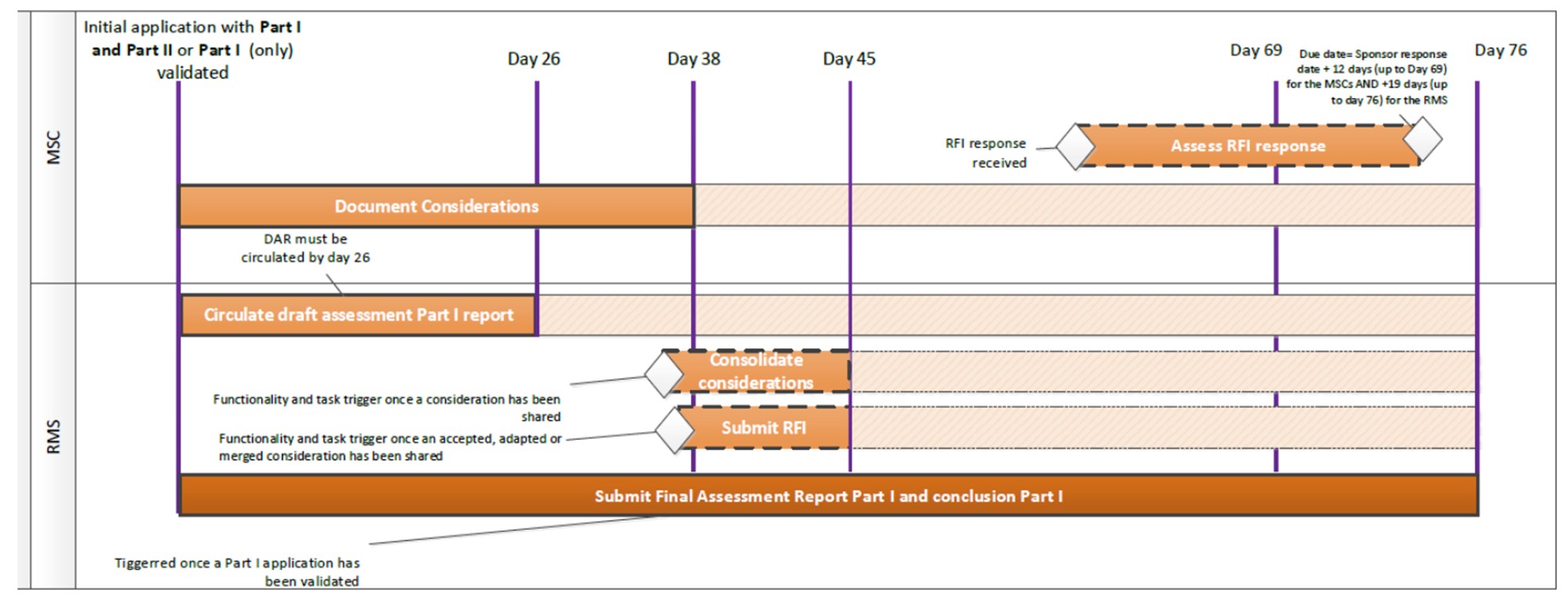{kind=link}
Vadovaujantis Lietuvos Respublikos biomedicininių tyrimų etikos įstatymo 242 straipsnio 1 dalimi, klinikinio vaistinio preparato tyrimo paraiškos 2014 m. balandžio 16 d. Europos Parlamento ir Tarybos reglamento (ES) Nr. 536/2014 dėl žmonėms skirtų vaistų klinikinių tyrimų, kuriuo panaikinama Direktyva 2001/20/EB, (toliau – Reglamentas (ES) Nr. 536/2014) 6 ir (ar) 7 straipsniuose nurodytais aspektais mokslinį vertinimą atlieka ir leidimą atlikti klinikinį vaistinio preparato tyrimą išduoda Valstybinė vaistų kontrolės tarnyba prie Sveikatos apsaugos ministerijos (toliau – Valstybinė vaistų kontrolės tarnyba), jeigu pagal šį vertinimą ir Lietuvos bioetikos komiteto klinikinio vaistinio preparato tyrimo paraiškos etinio vertinimo išvadą klinikinis vaistinio preparato tyrimas atitinka Reglamente (ES) Nr. 536/2014 nustatytus reikalavimus.
Vadovaujantis Lietuvos Respublikos biomedicininių tyrimų etikos įstatymo 242 straipsnio 2 dalimi, esminio klinikinio vaistinio preparato tyrimo pakeitimo paraiškos Reglamento (ES) Nr. 536/2014 6 ir (ar) 7 straipsniuose nurodytais aspektais mokslinį vertinimą atlieka ir leidimą atlikti klinikinio vaistinio preparato tyrimo pakeitimą išduoda Valstybinė vaistų kontrolės tarnyba, jeigu pagal šį vertinimą ir Lietuvos bioetikos komiteto esminio klinikinio vaistinio preparato tyrimo pakeitimo paraiškos etinio vertinimo išvadą klinikinio vaistinio preparato tyrimo pakeitimas atitinka Reglamente (ES) Nr. 536/2014 nustatytus reikalavimus.
Norint gauti leidimą atlikti klinikinį vaistinio preparato tyrimą arba leidimą atlikti esminius klinikinio vaistinio preparato tyrimo pakeitimus Lietuvos Respublikoje, klinikinio vaistinio preparato tyrimo užsakovas turi pateikti klinikinio vaistinio preparato tyrimo paraišką per Reglamento (ES) Nr. 536/2014 80 straipsnyje nurodytą ES portalą (toliau – ES portalas) Lietuvos Respublikai.
Pradinės paraiškos dokumentų rinkinys išsamiai aprašytas Reglamento (ES) Nr. 536/2014 I priede.
Esminio pakeitimo paraiškos dokumentų rinkinys išsamiai aprašytas Reglamento (ES) Nr. 536/2014 II priede.
Teikiant klinikinio vaistinio preparato tyrimo paraišką per ES portalą, galima naudoti Reglamento (ES) Nr. 536/2014 reikalavimus atitinkančius Europos Komisijos paskelbtų Taisyklių dėl vaistinių preparatų reglamentavimo Europos Sąjungoje 10 tome (angl. Eudralex. Volume 10. Clinical Trial Guidelines) skelbiamus dokumentų šablonus.
Informacija, kurią, vadovaudamiesi Reglamento (ES) Nr. 536/2014 reikalavimais, užsakovas ar jo įgaliotas atstovas turi pateikti Lietuvos Respublikai, norėdami gauti leidimą vykdyti klinikinį tyrimą, pateikti Reglamento (ES) Nr. 536/2014 I priede.
Siekdami suteikti užsakovams ir jų įgaliotiems atstovams aiškumo dėl Reglamente (ES) Nr. 536/2014 neapibrėžtų reikalavimų klinikinio vaistinio preparato tyrimo paraiškos II dalies dokumentų formai, pateikiame 2022 m. gegužės 17 d. Lietuvos bioetikos komiteto biomedicininių tyrimų ekspertų grupės posėdžio sprendimu patvirtintas Rekomendacijas dėl Lietuvos Respublikai teikiamų klinikinio vaistinio preparato tyrimo paraiškos II dalies dokumentų sąrašo ir pavyzdinių formų (toliau – Rekomendacijos).
Šiose Rekomendacijose išvardinti dokumentai turi būti pateikti lietuvių kalba.
Šių Rekomendacijų prieduose pateiktos pavyzdinės formos, skirtos užsakovams ir jų įgaliotiems atstovams, yra rekomenduojamos, tačiau neprivalomos naudoti.
I dalies dokumentų sąrašas:
- Lydraštis
- ES paraiška
- Protokolas
- Tyrėjo brošiūra
- Dokumentai, susiję su tiriamojo vaisto atitiktimi geros gamybos praktikai (jei tiriamasis vaistas yra registruotas, jų pateikti nereikia)
- Tiriamojo vaisto dokumentų rinkinys (TVDR) arba supaprastintas TVDR
- Pagalbinio vaisto dokumentų rinkinys
- Mokslinės rekomendacijas ir pediatrinių tyrimų planas (jei yra)
- Tiriamųjų vaistų ženklinimo turinys
II dalies dokumentų sąrašas:
10. Tiriamųjų įtraukimo į tyrimą ir informuoto asmens sutikimo procedūros aprašymas:
10.1. Tiriamųjų įtraukimo į tyrimą procedūros aprašymas (nuoroda (Word));
10.2. Reklaminių skelbimų kopijos, vaizdo ar garso įrašai (jeigu tiriamieji įtraukiami naudojant reklaminius skelbimus).
11. Informacija tiriamajam, asmens informavimo ir informuoto asmens sutikimo forma:
11.1. Rašytinė asmens informavimo ir informuoto asmens sutikimo forma (pavyzdinė forma) (rekomendacijos), (Rekomendacijos dėl tiriamųjų informavimo ir informuoto asmens sutikimo dalyvauti biomedicininiame tyrime davimo ir atšaukimo elektroninėmis priemonėmis (2024-07-16)), (Rekomendacijos. Vaikų informavimas apie dalyvavimą biomedicininiame tyrime)
11.2. Kita informacija, kuri bus pateikiama tiriamajam (pvz., tiriamojo kortelė).
12. Tyrėjo tinkamumą pagrindžiantys dokumentai:
12.1. Tyrimo centrų Lietuvoje sąrašas, kuriame nurodomos pagrindinių tyrėjų pavardės ir pareigos tyrime, planuojamas tiriamųjų skaičius juose (laisvos formos dokumentas);
12.2. Tyrėjų kvalifikacijos aprašymas ir patvirtinimas, kad tyrėjų kvalifikacija atitinka Europos Parlamento ir Tarybos reglamente (ES) Nr. 536/2014 ir Lietuvos Respublikos teisės aktuose nustatytiems reikalavimams (nuoroda (word));
12.3. Kiekvieno pagrindinio tyrėjo gyvenimo aprašymas (nuoroda (word));
12.4. Pagrindinių tyrėjų užpildytos interesų deklaracijos (nuoroda (word)).
13. Kiekvieno tyrimo centro vadovo ar jo įgalioto asmens rašytinis Tyrimo centro tinkamumo patvirtinimas (nuoroda (word)).
14. Civilinės atsakomybės draudimo liudijimas (polisas) arba kitas žalos atlyginimo užtikrinimo įrodymas.
15. Finansiniai ir kiti dokumentai:
15.1. Išlaidų kompensavimo tiriamiesiems už dalyvavimą tyrime aprašymas (nuoroda (word));
15.2. Tyrimo finansavimo, atlygio tyrėjams ir tyrimo centrui už dalyvavimą tyrime aprašymas bei kitų susitarimų tarp užsakovo ir tyrimo centro aprašymas (laisvos formos dokumentas).
16. Rinkliavos sumokėjimo patvirtinimas.
17. Užsakovo patvirtinimas, kad duomenys bus renkami ir tvarkomi vadovaujantis Bendruoju duomenų apsaugos reglamentu.
18. Informacija dėl atitikties valstybėje narėje taikomoms taisyklėmis dėl iš tiriamojo asmens paimtų biologinių mėginių rinkimo, saugojimo ir naudojimo ateityje (jeigu taikoma) (nuoroda (word)).
Valstybinė vaistų kontrolės tarnyba leidimą atlikti klinikinį vaistinio preparato tyrimą arba esminį klinikinio vaistinio preparato tyrimo pakeitimą išduoda ir Lietuvos bioetikos komitetas klinikinio vaistinio preparato tyrimo paraiškos arba esminio klinikinio vaistinio preparato tyrimo pakeitimo paraiškos etinio vertinimo išvadą pateikia Lietuvos Respublikos sveikatos apsaugos ministro 2022 m. sausio 27 d. įsakymu Nr. V-156 “Dėl Nacionalinio kontaktinio centro, nurodyto Europos Parlamento ir Tarybos reglamento (ES) Nr. 536/2014 dėl žmonėms skirtų vaistų klinikinių tyrimų, kuriuo panaikinama Direktyva 2001/20/EB, 83 straipsnyje, funkcijų vykdymo tvarkos aprašo patvirtinimo” patvirtinto Nacionalinio kontaktinio centro, nurodyto Europos Parlamento ir Tarybos reglamento (ES) Nr. 536/2014 dėl žmonėms skirtų vaistų klinikinių tyrimų, kuriuo panaikinama Direktyva 2001/20/EB, 83 straipsnyje, funkcijų vykdymo tvarkos apraše numatyta tvarka ir terminais.
Sprendimą dėl pritarimo klinikinio vaistinio preparato tyrimo paraiškos arba esminio klinikinio vaistinio preparato tyrimo pakeitimo paraiškos etinio vertinimo Reglamento (ES) Nr. 536/2014 straipsniuose nurodytais aspektais išvadai priima Lietuvos bioetikos komiteto biomedicininių tyrimų ekspertų grupė.
Už leidimo atlikti klinikinį vaistinio preparato tyrimą arba esminį klinikinio vaistinio preparato tyrimo pakeitimą imama nustatyto dydžio valstybės rinkliava.
- 2014 m. balandžio 16 d. Europos Parlamento ir Tarybos reglamentas (ES) Nr. 536/2014 dėl žmonėms skirtų vaistų klinikinių tyrimų, kuriuo panaikinama Direktyva 2001/20/EB. Nuoroda: https://eur-lex.europa.eu/legal-content/lt/TXT/?uri=CELEX%3A32014R0536.
- Lietuvos Respublikos biomedicininių tyrimų etikos įstatymas. Nuoroda: https://www.e-tar.lt/portal/lt/legalActEditions/TAR.234B15954C2F.
- Lietuvos Respublikos sveikatos apsaugos ministro 2022 m. sausio 27 d. įsakymas Nr. V-156 „Dėl Nacionalinio kontaktinio centro funkcijų, nurodytų Europos Parlamento ir Tarybos reglamento (ES) Nr. 536/2014 dėl žmonėms skirtų vaistų klinikinių tyrimų, kuriuo panaikinama Direktyva 2001/20/EB, 83 straipsnyje, vykdymo tvarkos aprašo patvirtinimo“. Nuoroda: https://www.e-tar.lt/portal/lt/legalAct/f3f082307f4111ec993ff5ca6e8ba60c.
- Lietuvos Respublikos sveikatos apsaugos ministro 2022 m. sausio 27 d. įsakymas Nr. V-157 „Dėl Geros klinikinės praktikos mokymų organizavimo tvarkos ir šių mokymų programų rengimo reikalavimų aprašo patvirtinimo“. Nuoroda: https://www.e-tar.lt/portal/lt/legalAct/c8e2c7b07f4111ec993ff5ca6e8ba60c.
Eudralex. Volume 10. Clinical Trial Guidelines. Nuoroda: https://ec.europa.eu/health/medicinal-products/eudralex/eudralex-volume-10_en#fragment1.
ES portalas. Nuoroda: https://euclinicaltrials.eu/home.
Atnaujinimo data: 2026-04-16