Authorisation of clinical trials on medicinal products according to the requirements of the Regulation (EU) No. 536/2014
Clinical trial on a medicinal product application evaluation scheme (LBEC)
{kind=link}
Clinical trial on a medicinal product application evaluation scheme (CTIS)
{kind=link}
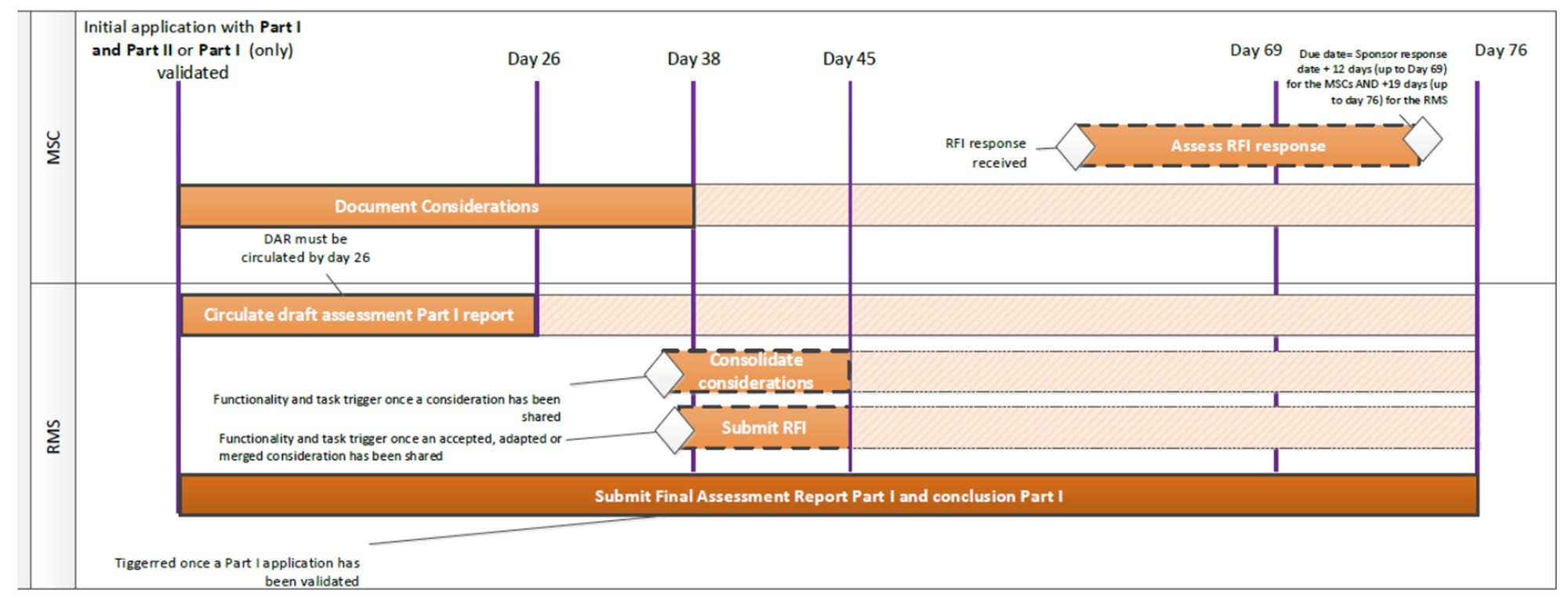
According to Article 242(1) of the Law on Ethics of Biomedical Research of the Republic of Lithuania (hereinafter – the Law), the scientific assessment of a clinical trial on a medicinal product shall be carried out and the authorization to conduct a clinical trial on a medicinal product shall be issued by the State Medicines Control Agency under the Ministry of Health (hereinafter - the Medicines Control Agency). The authorization shall be issued provided that, according to the assessment of the Medicines Control Agency and the conclusion of the ethical review of the Lithuanian Bioethics Committee, the clinical trial on a medicinal product complies with the requirements of the Regulation on Clinical Trials on Medicinal Products.
The decision on the approval of the draft conclusion of the ethical review shall be taken by the Group of the Biomedical Research Experts of the Lithuanian Bioethics Committee.
A sponsor wishing to conduct a clinical trial on a medicinal product in the Republic of Lithuania must submit a clinical trial application and other required documents to the EU portal referred to in Article 80 of Regulation on Clinical Trials on Medicinal Products.
The information that the sponsor or its authorized representative must submit to the EU portal in order to obtain a clinical trial authorization to conduct a clinical trial on a medicinal product in the Republic of Lithuania, is set out in Annex I of the Regulation on Clinical Trials on Medicinal Products.
List of Part I documents:
1. Cover letter
2. EU application
3. Protocol
4. Investigator's Brochure
5. Documents relating to the compliance of the IMP with good manufacturing practice (if the IMP is authorised, these are not required)
6. Investigational medicinal product dossier (IMPD) or simplified IMPD
7. Auxiliary medicinal product dossier
8. Scientific guidance and paediatric investigation plan (if applicable)
9. Content of the labelling of investigational medicinal products
In order to provide clarity to sponsors and their authorized representatives regarding the requirements of the Republic of Lithuania for the Part II documents of a clinical trial application, Lithuanian Bioethics Committee proposed the Recommendations on the List of Part II Documents and Templates (hereinafter – the Recommendations).
The Part II documents must be submitted in Lithuanian.
10. Description of the procedure for subject recruitment and informed consent:
10.1 description of the procedure for subject recruitment and obtaining informed consent (link (Word));
10.2. copies of advertisements, video or audio recordings (if subjects are recruited using advertisements).
11. Information for the subject, subject information and informed consent form:
11.1. written subject information and informed consent form (template LT) (recommendations ENG);
11.2. other information to be provided to the subject (e.g. subject card).
12. Documentation supporting suitability of the investigator:
12.1. a list of clinical trials sites in Lithuania, indicating the names and positions of the principal investigators and the number of subjects expected to be recruited;
12.2. Description of the qualification of the investigators and statement that qualification of the investigators meets the requirements listed in the Regulation (EU) No 536/2014 of the European Parliament and of the Council and in other legal documents of the Republic of Lithuania (link (Word));
12.3. a curriculum vitae of each principal investigator (link (Word));
12.4. completed declarations of interest signed by each principal investigator (link (Word)).
13. A written statement on the suitability of each clinical trial site issued by the head of the clinic/institution in the clinical trial site or his/her delegate (link (Word)).
14. Civil liability insurance certificate (policy) or other proof of indemnification.
15. Financial and other documents:
15.1. a description of the reimbursement of costs to subjects for participation in the clinical trial (link (Word));
15.2. a description of the financing of the clinical trial, information on financial transactions, compensation paid to subjects and investigator/site for participating in the clinical trial or any other agreement between the site.
16. Proof of payment.
17. A statement by the sponsor that the data will be collected and processed in accordance with the General Data Protection Regulation.
18. Information on compliance with Member State applicable rules for the collection, storage and future use of biological samples taken from the subject (if applicable) (link (Word)).
A state fee of a fixed amount shall be paid for the authorization of a clinical trial on a medicinal product or a substantial modification of a clinical trial on a medicinal product.
The application dossier for a substantial modification is detailed in Annex II of Regulation on Clinical Trials on Medicinal Products.
According to Article 242(2) of the Law, the scientific assessment of a substantial modification of a clinical trial on a medicinal product shall be carried out and the authorization to make a substantial modification of a clinical trial on a medicinal product shall be issued by the Medicines Control Agency. The authorization of a substantial modification shall be issued by the Medicines Control Agency upon the positive ethical assessment of the trial of the Group of the Biomedical Research Experts of the Lithuanian Bioethics Committee, that the substantial modification of a clinical trial on a medicinal product complies with the requirements of the Regulation on Clinical Trials on Medicinal Products.
The decision on the approval of the draft conclusion of the ethics review of a substantial modification shall be taken by the Group of the Biomedical Research Experts of the Lithuanian Bioethics Committee.
1. Eudralex. Volume 10. Clinical Trial Guidelines.
Reference: https://ec.europa.eu/health/medicinal-products/eudralex/eudralex-volume-10_en#fragment1.
2. EU Portal. Reference: https://euclinicaltrials.eu/home.
For more detailed information please contact:
Specialist Gita Labanauskaite, ph. +370 615 22703, e-mail: [email protected]